A seemingly innocuous lump can sometimes turn out to be a rare tumour, and thus should be considered with care. Although individual rare cancers have a very low incidence, they collectively account for 16-24% of cancer cases in East Asia – and present a significant diagnostic and therapeutic dilemma.
Early management of rare tumours at a specialised centre of care facilitates accurate diagnosis and treatment, which can otherwise be difficult without the joint expertise of an experienced multidisciplinary team. Prompt referral of such cases by general practitioners (GPs) and other surgical subspecialists is pivotal in improving patient outcomes.
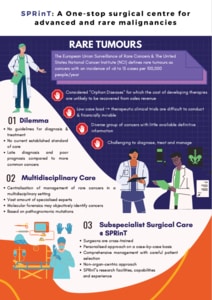
Figure 1 Rare tumours (click to expand infographic)
INTRODUCTION
Definition of rare cancers
Rare cancers have been defined by the European Union Surveillance of Rare Cancers in Europe (RARECARE) project and the United States National Cancer Institute (NCI) as cancers with an incidence of less than 6 to 15 cases per 100,000 people per year. 1-2
They comprise a diverse group of cancers with little available definitive information,3 and as such are challenging to diagnose, treat and manage.4-6
Clinical significance
Due to the low case load, clinical trials for rare cancer therapies are not only difficult to conduct4 but are also rarely financially viable. Along with other diseases of low prevalence, rare cancers are considered orphan diseases for which the cost of developing therapies is not likely to be recovered from sales revenue.7
These combined factors likely contribute to the poor prognosis associated with rare cancers compared to more common cancers.1,4
Despite their low individual incidence, the impact of rare cancers is significant as they collectively comprise a substantial proportion of the total cancer burden and mortality.8-9 Based on the RARECARE definition, the incidence of rare cancers as a percentage of all cancer diagnoses was 22% in Europe from 2000 to 2007, and 16 to 24% in East Asia (Japan, Korea and Taiwan) from 2011 to 2015.10
THE DIAGNOSTIC AND THERAPEUTIC DILEMMA
Rare cancers are challenging to manage as there are no guidelines for their diagnosis or treatment, and no current established standard of care.5
The Textbook of Uncommon Cancer proposes a general algorithm for management involving histopathological review by an experienced pathologist familiar with the site of origin, molecular and biochemical investigations, and radiological investigations. This analysis is best interpreted in centres of excellence, alongside a review of available literature as well as consultation with an expert with relevant publications.3
THE VALUE OF MULTIDISCIPLINARY CARE
In general, current literature favours the centralised management of rare cancers in a multidisciplinary setting,6 where a vast amount of specialised expertise is available from experienced surgical oncologists, medical oncologists, histopathologists and radiologists.
In the case of rare tumours, where diagnosis based on histopathology may be limited, molecular forensics may be required to more objectively identify cancers based on their pathognomonic mutations,9 which would likewise be more easily performed in specialised centres of care.
SUBSPECIALIST SURGICAL CARE AT SPRinT
At the Department of Sarcoma, Peritoneal and Rare Tumours (SPRinT) in the National Cancer Centre Singapore (NCCS), surgeons are cross-trained in various aspects of oncology with specific emphasis on tumour biology, technical competency and multi-visceral resection. They are also able to assimilate surgical options, either curative or palliative, in conjunction with other medical options or therapies offered by other subspecialties.
SPRinT offers a case-by-case, personalised approach; early, expert-guided management; and a non-organ-centric approach, which all have a positive impact on patient prognosis.11,12
Prior reviews have highlighted the importance of cooperative clinical and translational research efforts on the national and international scale in establishing evidence-based guidelines to diagnose and treat rare cancers.3-6 In view of the team’s capabilities and experience in coordinating national and international research initiatives, SPRinT is well poised to contribute to the improvement of future patient care and outcomes, in addition to a more comprehensive approach to patient care in the present.
CASE A
Rare presentation of myxoid epithelioid tumour of groin Presentation
Ms K is a 25-year-old lady who presented to a tertiary hospital with a left groin mass that had been increasing in size over the past two weeks. Ultrasound scan demonstrated a lobulated solid lesion measuring 6.9 x 6.2 x 1.4 cm with cystic areas within and internal vascularity. It was initially deemed to be not worrying, hence an excision biopsy was performed.
However, as histology was suggestive of a malignant myxoid epithelioid tumour and due to the rarity and difficulty in diagnosis of the tumour, she was referred to SPRinT (part of the expert multidisciplinary sarcoma board at NCCS) for a second opinion as well as management, follow-up and prognostication. She also had an overseas consultation. Multidisciplinary care: discussion, investigations and treatment Her case was discussed by the multidisciplinary tumour board involving pathologists and specialists in nuclear medicine, oncologic imaging, medical oncology, radiation oncology and surgical oncology. Review of the histology confirmed the diagnosis of a myxoid epithelial tumour, and further molecular forensics showed that it was a low grade malignant tumour with close margins.
Ms K underwent a wider excision of the excision biopsy site, with reconstruction of the defect involving a pedicled superficial circumflex iliac artery perforator flap, and was reassured of its indolent nature and likely good outcome. Outcomes
Ms K recovered well postoperatively. Due to the rarity and uncertain biology of the tumour, the SPRinT team is monitoring her case carefully with multidisciplinary involvement and will continue to manage it accordingly.
CASE B
Merkel cell tumour requiring multidisciplinary input and treatment Presentation
Mr O is a 77-year-old gentleman who presented to a tertiary institution with a right thigh skin nodule that had appeared a month ago. Biopsy revealed a rare malignant Merkel cell carcinoma, and he was referred to SPRinT for multidisciplinary discussion and treatment. Multidisciplinary care: discussion, investigations and treatment
Mr O underwent wide excision of the tumour with a skin graft. A year later, a new nodule was detected during follow-up which was shown to be recurrent Merkel cell carcinoma on punch biopsy. Further investigations showed no evidence of local or distal metastases. He underwent wider excisions with a split skin graft and sentinel lymph node biopsy.
Months later, an indeterminate nodule was found at the resection site. Mr O completed neoadjuvant radiotherapy as recommended following a multidisciplinary tumour board discussion.
Mr O is currently well and on regular follow-up with the SPRinT team. |
THE SPRinT SERIES: PARTNERING GPs TO CARE FOR ADVANCED AND RARE CANCERS
This is the fifth in a five-part series of articles, ‘The SPRinT Series’ – in which we discuss the work that SPRinT does, the key aspects of SPRinT’s clinical focus and the role of general practitioners (GPs) in providing care.
Due to the rarity of these tumours, it is critical for patients to be promptly referred to a specialist department/centre, such as SPRinT, to provide the best outcomes.
GPs are our first line of defence, and close collaboration between SPRinT and the GP community is essential for the timely diagnosis and referral of patients with rare tumours.
Read the other parts of the series here:
For GP referrals to the Department of Sarcoma, Peritoneal and Rare Tumours (SPRinT), please contact NCCS or the Patient Liaison Service (PLS) – GP Network at SGH at:
Hotline: 6436 8288
Email: SPRinT@singhealth.com.sg / gpnetwork@sgh.com.sg
Click here for more information on the department. |
REFERENCES
Gatta G, Capocaccia R, Trama A, Martínez-García C. The Burden of Rare Cancers in Europe. In: Posada de la Paz M, Groft SC, eds. Rare Diseases Epidemiology. Advances in Experimental Medicine and Biology. Springer Netherlands; 2010:285-303. doi:10.1007/978-90-481-9485-8_17 2.
Greenlee RT, Goodman MT, Lynch CF, Platz CE, Havener LA, Howe HL. The Occurrence of Rare Cancers in U.S. Adults, 1995–2004. Public Health Rep. 2010;125(1):28-43. doi:10.1177/003335491012500106
Raghavan D. A Structured Approach to Uncommon Cancers. In: Textbook of Uncommon Cancer. John Wiley & Sons, Ltd; 2017:1-5. doi:10.1002/9781119196235.ch1
Mittra A, Takebe N, Florou V, Chen AP, Naqash AR. The emerging landscape of immune checkpoint inhibitor based clinical trials in adults with advanced rare tumors. Hum Vaccines Immunother. 2021;17(7):1935-1939. doi:10.1080/21645515.2020.1854604
Mathoulin-Pélissier S, Pritchard-Jones K. Evidence-based data and rare cancers: The need for a new methodological approach in research and investigation. Eur J Surg Oncol J Eur Soc Surg Oncol Br Assoc Surg Oncol. 2019;45(1):22-30. doi:10.1016/j.ejso.2018.02.015
Gatta G, Capocaccia R, Botta L, et al. Burden and centralised treatment in Europe of rare tumours: results of RARECAREnet—a population-based study. Lancet Oncol. 2017;18(8):1022-1039. doi:10.1016/S1470-2045(17)30445-X
Commissioner O of the. Orphan Drug Act - Relevant Excerpts. FDA. Published online April 24, 2019. Accessed April 15, 2022. https://www.fda.gov/industry/designating-orphan-product-drugs-and-biological-products/orphan-drug-act-relevant-excerpts
Hong AL, Tseng YY, Cowley GS, et al. Integrated genetic and pharmacologic interrogation of rare cancers. Nat Commun. 2016;7(1):11987. doi:10.1038/ncomms11987
Boyd N, Dancey JE, Gilks CB, Huntsman DG. Rare cancers: a sea of opportunity. Lancet Oncol. 2016;17(2):e52-e61. doi:10.1016/S1470-2045(15)00386-1
Matsuda T, Won YJ, Chun-ju Chiang R, et al. Rare cancers are not rare in Asia as well: The rare cancer burden in East Asia. Cancer Epidemiol. 2020;67:101702. doi:10.1016/j.canep.2020.101702
Tu IWH, Wong JSM, Tan QX, Ng G, Ong CAJ, Chia CS. Department of Sarcoma, Peritoneal and Rare Tumors (SPRinT): A subspecialty surgical oncological care model for advanced malignancies requiring complex procedures. Asian J Surg. 2022;45(1):546-548. doi:10.1016/j.asjsur.2021.09.018
Bilimoria KY, Phillips JD, Rock CE, Hayman A, Prystowsky JB, Bentrem DJ. Effect of surgeon training, specialization, and experience on outcomes for cancer surgery: a systematic review of the literature. Ann Surg Oncol. 2009;16(7):1799-1808. doi:10.1245/s10434-009-0467-8
Tags:
;
;
;
;
News Article;
National Cancer Centre Singapore;
National Cancer Centre Singapore;
Article;
MedAdvance;
;
;
Singapore General Hospital;Sarcoma, Peritoneal and Rare Tumours (SPRinT);
;
MedAdvance;Patient Care

